7月10日����,國家藥品監督管理局發(fā)布了《接受藥品境外臨床試驗數據的技術(shù)指導原則》(下稱(chēng)《原則》)���。這意味著(zhù)���,國家食藥監總局藥審中心在2017年10月20日發(fā)布的《接受境外臨床試驗數據的技術(shù)要求》征求意見(jiàn)稿采納了合理意見(jiàn)建議后的正式落地�����。
去年年底����,即征求意見(jiàn)稿出臺不久�,中國醫學(xué)科學(xué)院藥物研究所新藥開(kāi)發(fā)研究室主任�����、國家食品藥品監督管理總局新藥審評委員吳松在接受《人民日報》采訪(fǎng)時(shí)表示����,“共享”臨床數據將極大減少進(jìn)口新藥在中國獲批上市的時(shí)間����,同時(shí)也有利于中國醫藥企業(yè)走向海外����,開(kāi)展境外臨床試驗�����。
接受境外臨床數據到底利好哪些企業(yè)呢�?筆者在與行業(yè)人士交流后發(fā)現��,大家普遍認為利好的企業(yè)至少包括以下5類(lèi):(1)跨國大藥企����;(2)從海外引進(jìn)在研項目的創(chuàng )新藥企��;(3)率先在美國���、澳大利亞等境外開(kāi)展臨床試驗的創(chuàng )新藥企�����;(4)在印度等境外開(kāi)展仿制藥BE試驗的企業(yè)����;(5)開(kāi)發(fā)危重疾病���、罕見(jiàn)病����、兒科的企業(yè)��。
跨國大藥企
過(guò)去����,一些新藥在我國的上市時(shí)間�����,平均要比歐美晚5到7年����。當時(shí)部分原因是��,中國并不認可在境外多中心取得的臨床試驗數據��,國外新藥首次進(jìn)入中國�,必須在國外做完Ⅱ期臨床試驗后才能在中國進(jìn)行臨床試驗�,在中國完成Ⅲ期臨床試驗后才能獲批上市�����。
《原則》明確提出:只要是真實(shí)可靠�����、符合ICH GCP和藥品注冊檢查要求�����;支持目標適應癥的有效性和安全性評價(jià)�����;不存在影響有效性和安全性的種族敏感性因素的境外臨床研究數據都能夠被CDE完全接受�����。即使是存在影響有效性和/或安全性的種族敏感性因素�����,數據外推至中國人群的有效性和安全性評價(jià)存在較大的不確定性�����,CDE也可以部分接受���。
這顯然讓“有條件”接受境外臨床數據更明晰���,從機制上縮短境外新藥在國內上市的時(shí)間���,畢竟數據可以在一定程度上通用了���,患者或許也能以最快速度享受到優(yōu)質(zhì)的創(chuàng )新藥品�����,而不用像以往一樣等上數年或通過(guò)非正式渠道從境外獲取“救命藥”�。
從海外引進(jìn)在研項目的創(chuàng )新藥企
近年來(lái)中國藥企研發(fā)和資金實(shí)力逐漸增強��,在國家鼓勵創(chuàng )新的政策引導下�,國內企業(yè)越來(lái)越多地與跨國企業(yè)建立合作���,通過(guò)專(zhuān)利許可�、共享權益等方式引入國際創(chuàng )新研發(fā)項目�。
一方面�����,這種通過(guò)跨境合作引進(jìn)的方式比自主研發(fā)更快捷�,國內藥企也是希望借助與海外企業(yè)合作的方式提升國際化研發(fā)能力���。另一方面�����,跨國大藥企有聚焦核心業(yè)務(wù)����、剝離非核心研發(fā)項目資產(chǎn)的考慮�,海外中小型生物醫藥公司則是希望與本土有研發(fā)實(shí)力的藥企合作來(lái)快速拓展中國市場(chǎng)��。
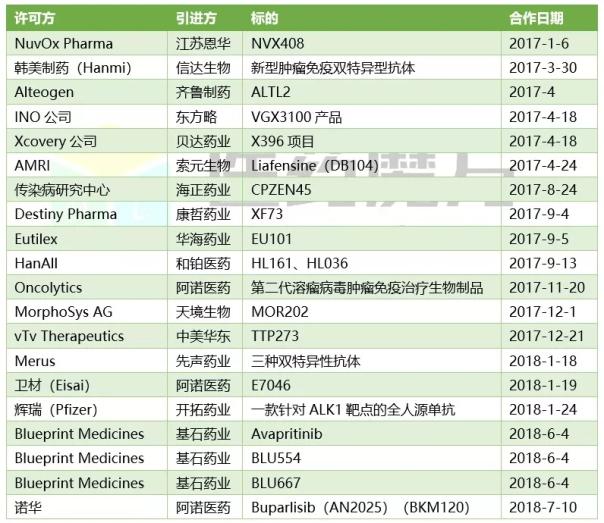
中國創(chuàng )新藥海外引進(jìn)案例(部分)
在剛剛過(guò)去的6月����,基石藥業(yè)一舉從美國2家中型生物技術(shù)公司拿下商業(yè)化引進(jìn)4個(gè)項目����。該公司商務(wù)拓展負責人告訴記者�,美國中小型生物技術(shù)并不具備在亞太地區商業(yè)化開(kāi)發(fā)能力�,他們通過(guò)與國內具有臨床開(kāi)發(fā)能力的創(chuàng )新藥企合作���,推動(dòng)授權產(chǎn)品在大中華區的單藥及聯(lián)合治療的臨床開(kāi)發(fā)和商業(yè)化����。CDE認可境外數據�����,或將進(jìn)一步加速產(chǎn)品在中國的上市進(jìn)程���。
在境外開(kāi)展臨床試驗再回國內申報的企業(yè)
國內創(chuàng )新藥開(kāi)發(fā)企業(yè)在藥品注冊時(shí)一般采取兩類(lèi)策略:一是獲得國家藥監局臨床批件后���,在國內開(kāi)展臨床試驗���,結束后申報新藥并在國內上市�����;二是同步開(kāi)展中國和國外的臨床研發(fā)���,待試驗結束后分別申請中國及國外新藥批文�,在全球同步上市��。
一位新藥開(kāi)發(fā)負責人向記者透露:較高的研究標準和質(zhì)量標準(特別是早期階段的能力)�、有可比性的成本�����、及時(shí)的審判批準和可靠的病人招募等因素使得澳大利亞成為極具吸引力的臨床試驗國家���。即便是同步準備IND申報資料��,在澳大利亞的推進(jìn)速度可能會(huì )更快��。
《原則》指出:鼓勵開(kāi)展境內外同步研發(fā)�����。境外臨床試驗數據可以用于支持需要進(jìn)行有效性和/或安全性評價(jià)的各類(lèi)注冊申請��。例如��,在境外開(kāi)展的早期臨床研究數據����,可用于支持在我國開(kāi)展注冊臨床試驗的申請��。這對于創(chuàng )新藥開(kāi)發(fā)企業(yè)而言�����,顯然是降低了開(kāi)發(fā)成本����。
鴻運華寧是一家十年如一日堅守開(kāi)發(fā)原創(chuàng )性新藥的企業(yè)��。該公司新藥開(kāi)發(fā)負責人告訴記者���,創(chuàng )新藥在澳洲開(kāi)展床新藥的I期臨床����,具有快速����、花費少��、質(zhì)量高的特點(diǎn)�����;申辦方可降低研發(fā)成本�,縮短研發(fā)周期���,提高成功率����。不過(guò)他也坦言�,在健康受試者身上的試驗優(yōu)勢����,在后期進(jìn)行患者試驗時(shí)����,會(huì )受到患者資源的限制��。國家藥監總局出臺的新政���,若能讓創(chuàng )新藥企借力于澳大利亞等國家的早期開(kāi)發(fā)數據���,支持后續在國內的注冊申報��,將會(huì )大大節省研發(fā)資源��,避免重復浪費��。
在境外開(kāi)展仿制藥BE試驗的企業(yè)
在國家藥品監督管理局發(fā)布的《接受藥品境外臨床試驗數據的技術(shù)指導原則》解讀中明確指出:境外完成的仿制藥生物等效性(BE)試驗數據����,研究質(zhì)量好�����,具備真實(shí)性�、完整性����、準確性和可溯源性的�,也可用于在我國的注冊申請����。
據了解����,在過(guò)去���,國內制劑企業(yè)在出口美國���、歐盟時(shí)���,通過(guò)在印度等境外國家進(jìn)行預BE/BE試驗�,不僅可以降低成本�����,還能提高數據被FDA/歐盟的認可度�����。一般這些“出?����!碑a(chǎn)品被歐美批準后����,再通過(guò)海外共線(xiàn)生產(chǎn)轉報國內的“曲線(xiàn)回國”方式在國內上市�����。醫藥魔方記者為此也向行業(yè)內相關(guān)人士求證��。
上海安必生制藥董事長(cháng)雷繼峰在接受記者采訪(fǎng)時(shí)表示:仿制藥生物等效試驗(BE)是比較兩個(gè)處方在同一個(gè)人體(受試者)上的體內表現�����。BE試驗結果與受試者人種種族無(wú)關(guān)��,與處方工藝��、BE試驗設計有一定關(guān)系�,歐美藥監機構和WHO專(zhuān)家對此認識完全一致���。歐美和WHO均接受世界任何國家符合GCP和GLP的BE數據�����。因此向FDA申報仿制藥申請(ANDA)����,沒(méi)有硬性規定必須在美國境內做BE���,也可以在加拿大��、印度等境外國家做�,前提是只要符合GLP和GCP管理的臨床試驗基地����,同時(shí)數據符合ICH要求����。中國加入了ICH后���,也同樣要求在數據上與國際接軌�����。
曾經(jīng)與印度CRO公司合作過(guò)100多項BE試驗的雷繼峰還透露���,印度CRO產(chǎn)業(yè)高度發(fā)展��,已形成規?���;?����,大部分CRO公司能夠像歐美公司一樣�,可以提供BE試驗一站式服務(wù)����。不過(guò)���,由于印度受試者群體大��,能加快入組速度�;同時(shí)相比于歐美甚至中國��,印度的BE服務(wù)費用更加低廉�����,是吸引越來(lái)越多藥企到印度做BE的主要原因�����。據不完全統計�,在向FDA申請ANDA的項目中�,每年約有50%到60%是在印度做BE����。
醫藥魔方記者獲悉��,早在兩年前�,泰格醫藥已經(jīng)在印度成立子公司(見(jiàn):泰格醫藥在印度成立子公司�����,提高臨床試驗數據管理���、統計分析及醫學(xué)寫(xiě)作能力)���。一位仿制藥開(kāi)發(fā)的資深專(zhuān)業(yè)人士也向記者證實(shí):除了FDA���,還有歐盟都會(huì )認可在印度等境外國家的臨床試驗數據�����,只要數據真實(shí)性����、完整性�、準確性和可溯源性等符合ICH要求�。如今��,CDE直接認可印度的BE數據�,顯然降低了開(kāi)發(fā)成本�,也省去了企業(yè)“曲線(xiàn)回國”的路徑�,于國內因一致性評價(jià)“坐地漲價(jià)”的CRO企業(yè)而言���,這一政策或帶來(lái)一定打擊����。
雷繼峰坦言����,我國認可仿制藥的境外BE數據對制藥行業(yè)本身而言是好事��,這不僅有利于國內藥企降低臨床開(kāi)發(fā)成本��;同時(shí)也能幫助老百姓早日吃上價(jià)格親民的仿制藥�;盡管短期內對國外CRO有些鎮痛����,但長(cháng)遠看是有利于國內CRO學(xué)習國際規范�����,提高專(zhuān)業(yè)水平�,并打破行業(yè)資源壟斷����。
開(kāi)發(fā)危重疾病��、罕見(jiàn)病����、兒科的企業(yè)
《原則》在最后還特別提到��,對于用于危重疾病�、罕見(jiàn)病��、兒科且缺乏有效治療手段的藥品注冊申請��,屬于“部分接受”情形的���,可有條件接受�����。
以開(kāi)發(fā)“罕見(jiàn)病”治療的孤兒藥為例�����。此前上市的明星藥物西達本胺是一款用于治療復發(fā)或難治性外周T細胞淋巴瘤的罕見(jiàn)病藥物��。生產(chǎn)商微芯生物總裁兼首席科學(xué)官魯先平曾在接受媒體采訪(fǎng)時(shí)表示��,由于公司與國家食藥監總局藥品審評中心溝通充分�,西達本胺的審評審批速度得以加快����。但即便如此����,微芯生物也花了4年時(shí)間才完成了近100例病人的二期試驗��。到2015年西達本胺獲準上市時(shí)�,該藥20年的專(zhuān)利獨占期僅剩8年�����。
國家藥監總局出臺的新政提出對“部分接受”情形有條件接受�����,對那些海外臨床數據“不完美”的孤兒藥產(chǎn)品���,有望在國內“有條件批準”上市����。醫藥前不久也做了統計調研�����,越來(lái)越多的國內創(chuàng )新藥企借力孤兒藥政策�,實(shí)現彎道超車(chē)�����。(見(jiàn):這13個(gè)國產(chǎn)新藥�,獲得了FDA孤兒藥資格認定)�。種種跡象表明����,這顯然是利好孤兒藥開(kāi)發(fā)企業(yè)的����。
獲得FDA孤兒藥資格認定的國產(chǎn)創(chuàng )新藥